Software Tools and Ecosystem
The Evolution of the Volume Electron Microscopy Pipeline
The advent of Serial Block-Face Scanning Electron Microscopy (SBF-SEM) and other volume electron microscopy (vEM) modalities has driven a "quiet revolution" in biological imaging [9, 10]. By enabling the automated acquisition of continuous, high-resolution ultrastructural data across three dimensions, SBF-SEM bridges the gap between traditional 2D transmission electron microscopy (TEM) and broader tissue-level imaging [3, 17, 46]. However, as the throughput of modern electron microscopes has increased and sample preparation protocols have advanced, the primary bottleneck in vEM has decisively shifted away from data acquisition [14, 16]. The automated, slice-and-view methodology of SBF-SEM generates massive datasets routinely comprising hundreds to thousands of serial images, often equating to tens or hundreds of gigabytes—or even terabytes—of raw data per specimen [17, 67, 92, 149, 256].
Managing, processing, and extracting meaningful biological insights from these enormous datasets necessitates a highly specialized software ecosystem [35]. The computational workflow for SBF-SEM generally follows a linear progression: automated microscope control and image acquisition, data handling and spatial alignment, image segmentation (both manual and automated), and finally, three-dimensional rendering and quantitative spatial analysis [14, 19]. To address these challenges, the field relies on a combination of proprietary, vendor-supplied applications and an expansive library of open-source software packages that have been developed and refined by the community [17, 35].
Acquisition Control and System Automation
The initial stage of the SBF-SEM workflow involves the precise coordination of the scanning electron microscope (SEM) hardware, the backscattered electron (BSE) detector, and the in-chamber ultramicrotome [19, 27, 74]. Commercial SBF-SEM systems are typically bundled with proprietary software, such as Gatan’s DigitalMicrograph (for the 3View system) or Zeiss’s SmartSEM, which manage basic imaging parameters such as accelerating voltage, dwell time, and slice thickness [17, 74, 127]. While these standard applications are sufficient for fundamental imaging, they often lack the flexibility, speed, and advanced error-handling capabilities required for continuous, multi-day acquisitions of complex biological volumes [74]. For example, image acquisition via traditional scan generators can be artificially rate-limited, and long runs are highly susceptible to sudden hardware drift, charging artifacts, or debris falling onto the block face [50, 74] (Figure 61).
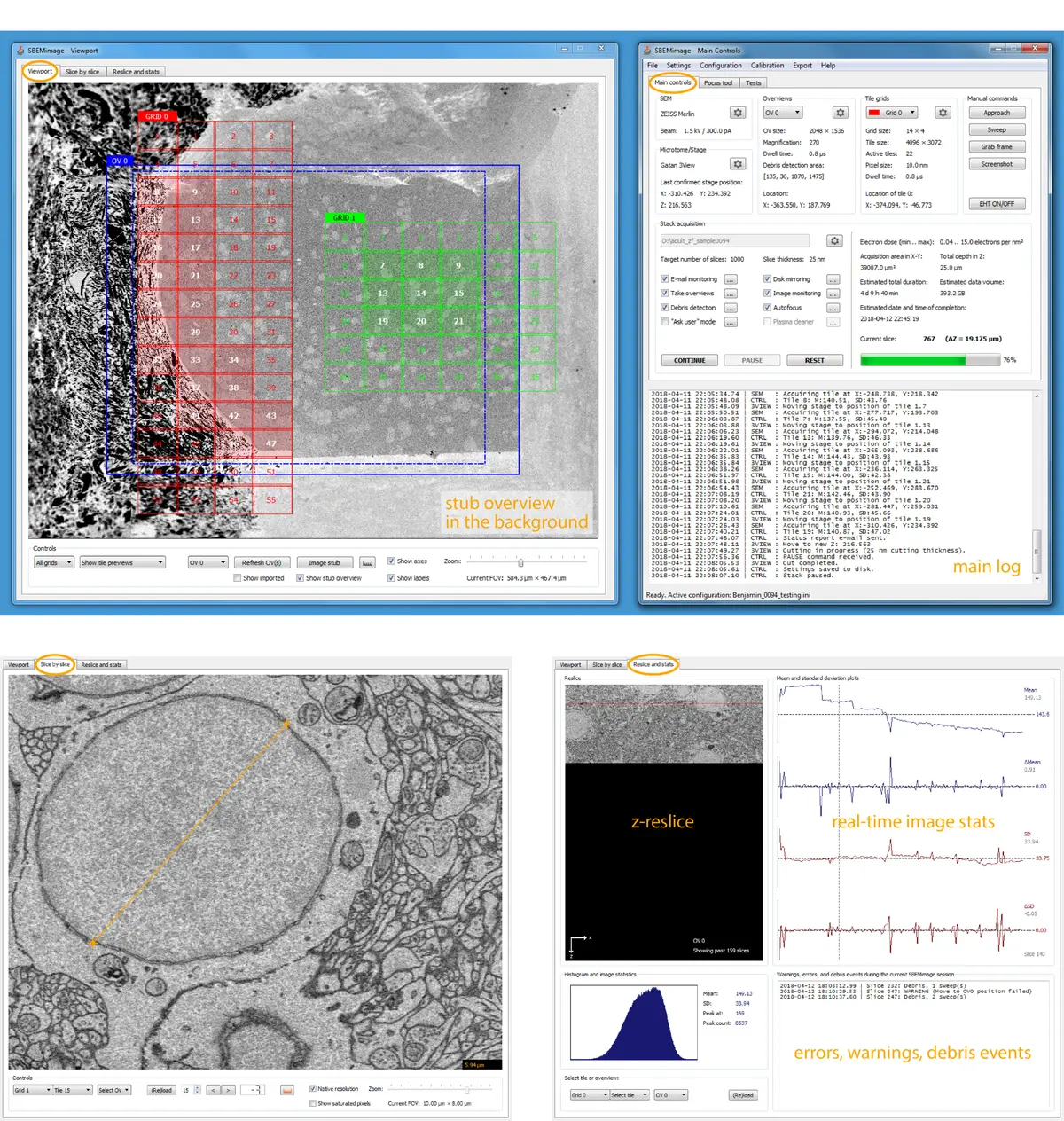
To overcome these limitations, researchers have developed specialized, open-source acquisition control software. A prominent example is SBEMimage, a Python-based application designed to execute complex SBF-SEM acquisitions by interacting directly with the SEM’s low-level proprietary APIs [74]. SBEMimage facilitates enhanced acquisition speeds and provides crucial automated quality control features tailored for long-term stability [74]. Chief among these is automated debris detection; the software can analyze pixel variations in defined quadrants of the block face to identify occluding debris, subsequently triggering the microtome to perform a "sweep" to clear the surface before resuming imaging [74].
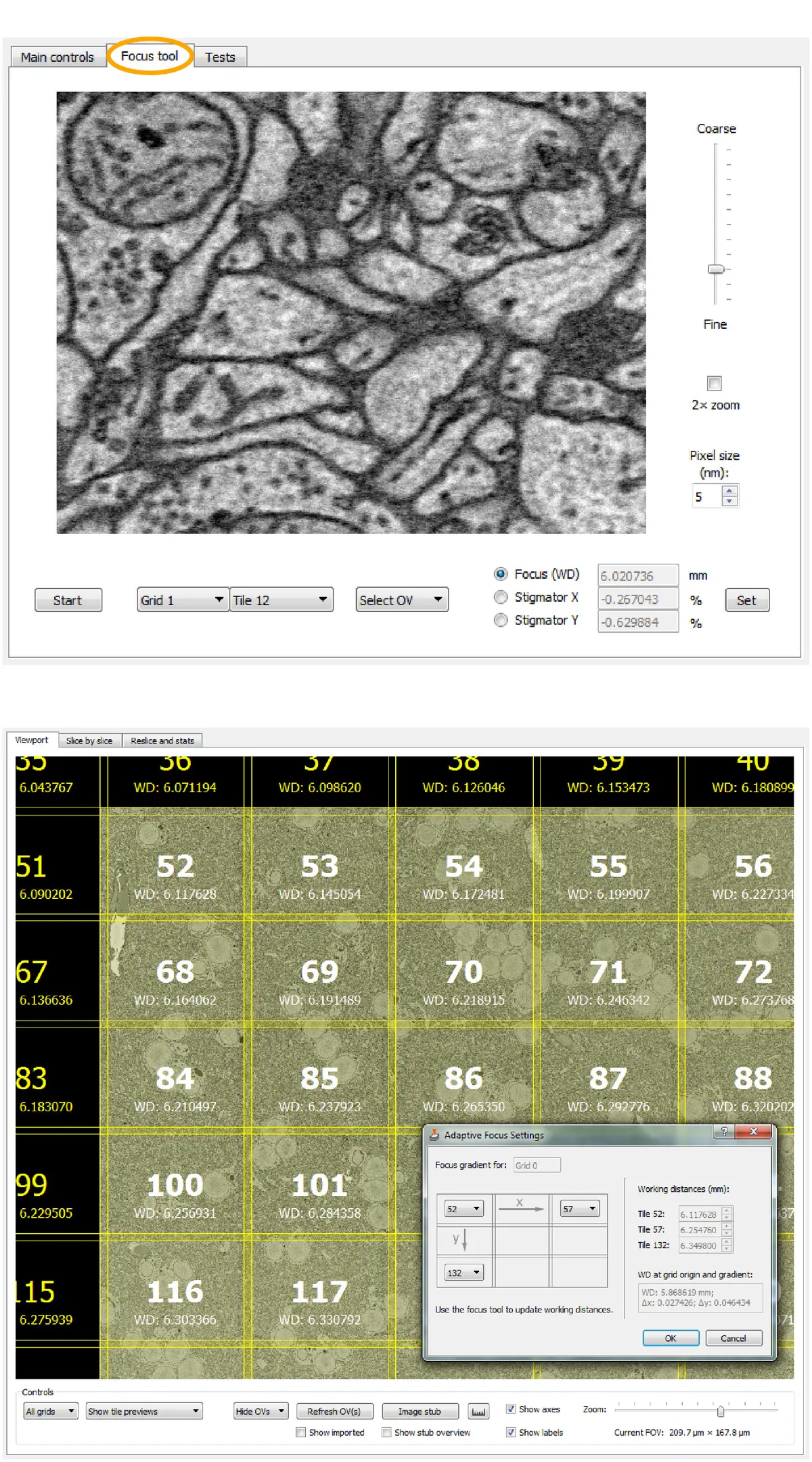
Furthermore, advanced control software allows for sophisticated region-of-interest (ROI) management. SBEMimage features adaptive tile selection, which permits users to define complex, non-rectangular imaging grids that follow the specific contours of a tissue sample (such as a whole organism or a specific neural tract), drastically reducing unnecessary data acquisition and storage requirements [74]. Integrated tools for remote autofocus, focus gradient correction across tilted surfaces, and slice-by-slice tile monitoring further ensure that unexpected changes in image quality automatically pause the system and alert the user, preventing catastrophic data loss [74]. Other bespoke acquisition platforms, such as piTEAM for multibeam systems or WaferMapper for array tomography, similarly demonstrate the community's push toward highly automated, data-driven acquisition frameworks [9]. Recent commercial integrations, such as the TESCAN 3D Analysis Suite paired with ConnectomX microtomes, also natively incorporate SBEMimage, indicating a growing convergence between hardware vendors and open-source control ecosystems [19] (Figure 62).
Data Handling, Pre-Processing, and Alignment
Following acquisition, the raw image data—often output in proprietary formats like .dm3 or .dm4—must be converted into accessible stack formats (e.g., .tiff or .mrc) for downstream processing [17, 18, 81, 111]. SBF-SEM data is inherently well-registered compared to serial section TEM or array tomography because the sample block remains fixed relative to the electron beam and detector [14, 81]. However, minor spatial misalignments frequently occur due to sample charging, thermal drift, or minute mechanical vibrations [28, 111]. Restoring absolute 3D continuity to the serial image stack is a prerequisite for accurate biological segmentation [14, 275].
The open-source platform FIJI/ImageJ serves as the foundational hub for SBF-SEM pre-processing [17, 35, 81, 161]. To correct stage drift, users commonly employ the Scale Invariant Feature Transform (SIFT) algorithm plugin, which globally minimizes registration errors by detecting and matching local features across adjacent slices [5, 9, 28, 81, 111]. For datasets involving expansive tiled mosaics, the TrakEM2 plugin within FIJI provides robust elastic and rigid image registration capabilities, allowing for the seamless stitching and alignment of massive fields of view [9, 55, 161, 164]. Other dedicated alignment tools, such as the Alignment to Median Smoothed Template (AMST) algorithm and automated stitching pipelines like vEMstitch, have been developed to correct local pixel variations and resolve boundary ghosting in highly heterogeneous samples [9, 276].
Preprocessing also involves image enhancement to mitigate the inherent noise of low-dose electron imaging. SBF-SEM stacks are routinely subjected to Gaussian blurring, unsharp masking, contrast normalization, and anisotropic filtering to improve the signal-to-noise ratio [50, 81, 156, 277]. More advanced approaches employ Total Variation (TV) or the ROF (Rudin-Osher-Fatemi) model for denoising while preserving distinct structural edges [88]. Additionally, computational deconvolution—historically restricted to light microscopy—is now being applied to SBF-SEM. By modeling the 3D point spread function (PSF) of electron scattering within the resin, software like Huygens can mathematically deconvolve the dataset, significantly improving z-axis resolution and overall data quality [145].
Given the immense size of these datasets, loading entire volumes into standard RAM is often impossible [9, 164]. To circumvent this, the software ecosystem has embraced virtual stacks and chunk-based multi-scale image pyramids [9, 28, 111]. Tools like BigDataViewer and the MoBIE plugin within FIJI, BigDataProcessor2, and the Python-based *napari* viewer enable the rapid out-of-core loading and fluid exploration of terabyte-scale volumes on standard local workstations [9].
Image Segmentation: From Manual Tracing to Machine Learning
The most significant computational bottleneck in the SBF-SEM pipeline is image segmentation—the process of assigning pixels to specific biological structures (e.g., organelles, membranes, or cells) to generate a 3D model [14, 18, 28]. Because cellular structures in vEM are identified primarily by their heavy metal contrast and complex morphology within a highly crowded intracellular environment, automated segmentation remains a formidable computer vision challenge [9, 87, 167].
#### Manual and Semi-Automated Tools For precise ground-truth generation or the analysis of rare morphological events, researchers rely on manual and semi-automated segmentation suites [9, 167]. Programs such as IMOD, Amira/Avizo, Microscopy Image Browser (MIB), and ORS Dragonfly are the mainstays of this process [28, 35, 81].
IMOD (specifically the *etomo* and *3dmod* components) has a long history in electron tomography but is heavily utilized in SBF-SEM for its robust drawing tools, allowing users to manually trace cross-sectional contours that are subsequently meshed into 3D volumes [23, 38, 81, 111, 121, 136, 280]. Microscopy Image Browser (MIB) is a highly versatile, open-source MATLAB-based platform specifically tailored for vEM [9, 28, 35]. MIB integrates manual annotation with powerful semi-automated features like local thresholding, morphological region-growing, and interpolation, bridging the gap between manual tracing and full automation [9, 28, 29, 155].
Commercial platforms offer highly optimized, GPU-accelerated environments with intuitive graphical user interfaces. Amira (Thermo Fisher) and Imaris (Bitplane) provide sophisticated semi-automated tools [35, 92, 247]. Amira’s "magic wand" and "blow tool" algorithms utilize contrast gradients and pixel densities to rapidly expand polygons and highlight contiguous structures like mitochondria or heavily stained lipid droplets [97, 281]. Similarly, Otsu’s thresholding method is frequently applied to separate bimodal signal distributions, isolating highly electron-dense structures (e.g., collagen networks or condensed chromatin) from the surrounding resin matrix [137, 277, 283].
#### Machine Learning and Deep Learning Implementations The sheer volume of SBF-SEM data renders purely manual segmentation intractable for large-scale connectomics or holistic cellular mapping [14, 18, 164]. Consequently, the field has rapidly integrated machine learning (ML) and deep learning (DL) into the software ecosystem [18, 167].
The interactive learning toolkit *ilastik* is widely used for its Random Forest pixel classification [9, 28, 179]. By providing minimal user annotations (scribbles) denoting foreground and background, *ilastik* trains a classifier in real-time, enabling the rapid extraction of complex organelles like the endoplasmic reticulum or synaptic vesicles across thousands of slices [28, 88].
More recently, Convolutional Neural Networks (CNNs) and deep learning architectures like U-Net have achieved state-of-the-art segmentation accuracy [9, 149, 167]. Initiatives such as the CellMap project have generated diverse, high-resolution ground-truth datasets to train generalizable deep neural networks capable of predicting boundary distances for dozens of subcellular organelles [167]. The Volume Segmentation Tool (VST) exemplifies the push to democratize these algorithms; designed to run entirely on local hardware with a browser-based GUI, VST automates data augmentation and model training for non-coding experts [149]. VST evaluates the 3D volume simultaneously rather than slice-by-slice, predicting contour maps to achieve both semantic (pixel-class) and instance (object-level) segmentation [149].
To address domain shift—where networks trained on one tissue fail on another due to variations in staining or voxel size—researchers increasingly employ transfer learning and domain adaptation algorithms [163]. Furthermore, innovative deep learning models like IsoVEM utilize video transformers and self-supervised strategies to computationally repair damaged slices and reconstruct highly isotropic data from fundamentally anisotropic SBF-SEM axial sampling [143].
3D Visualization, Rendering, and Quantitative Analysis
Transforming a segmented stack of 2D labels into a scientifically interpretable 3D model requires advanced visualization and analytical software [18, 19]. Extracted segmentations are algorithmically converted into triangulated surface meshes or rendered volumetrically based on voxel densities [12, 23, 97, 277].
Amira, ORS Dragonfly, and TESCAN's 3D Analysis Suite excel in generating smooth, high-fidelity surface renderings [19, 97]. These programs are deeply integrated with quantitative morphometric pipelines. Using modules like Amira's "Label Analysis" or IMOD's `imodinfo`, researchers can instantly extract localized geometric data, including individual organelle volumes, surface areas, spatial coordinates, branching tortuosity, and nearest-neighbor distances [18, 63, 121, 136, 247, 282]. Advanced geometric characterizations, such as calculating the form factor of segmented chromatin [283], or applying skeletonization algorithms (via FIJI's MorphoLibJ or Python) to determine absolute neurite lengths, provide the essential metrics for testing biological hypotheses [9, 136, 283].
For publication-quality visualization and cinematic animation, open-source 3D computer graphics software like Blender is increasingly integrated into the vEM pipeline [17, 35, 155, 211]. Add-ons such as NeuroMorph facilitate the direct import of segmented meshes into Blender, enabling customized lighting, complex camera trajectories, and the export of models to interactive web platforms like Sketchfab [155, 211].
Additionally, vEM datasets are frequently coregistered with fluorescence light microscopy in Correlative Light and Volume Electron Microscopy (vCLEM) workflows [3, 46, 99]. Software like CLEM-Reg, distributed as a plugin for the *napari* viewer, utilizes machine-learning-derived point clouds (often anchored by segmented mitochondria) to automate the highly complex spatial registration of differing imaging modalities, negating the need for manual landmark placement [124].
Collaborative Annotation and Web-Based Ecosystems
Because vEM projects (particularly in neuroanatomy and connectomics) encompass volumes that exceed the analytical capacity of a single laboratory, collaborative, web-based software ecosystems have become indispensable [9, 46, 164]. Rather than passing terabyte-sized hard drives between institutions, data is hosted on centralized servers and streamed to end-users on demand [9].
Platforms such as *webKnossos*, *CATMAID* (Collaborative Annotation Toolkit for Massive Amounts of Image Data), and Google's *Neuroglancer* allow globally distributed teams to view, annotate, and proofread vEM data concurrently via standard web browsers [9]. The Knossos family of tools provides specialized environments for the rapid manual skeletonization and node-tracing of neuronal arbors [9, 17]. This server-based approach has also enabled massive crowdsourcing initiatives; platforms like *EyeWire* successfully gamify the segmentation process, harnessing citizen scientists to map retinal connectomes [17].
To ensure the reproducibility, reuse, and long-term utility of these computationally expensive datasets, the community is establishing rigorous metadata standards [9]. Guidelines developed by consortia such as QUAREP-LiMi and the Recommended Metadata for Biological Images (REMBI) ensure that critical parameters regarding sample preparation, SBF-SEM acquisition, and software segmentation pipelines are permanently linked to the primary image data in public repositories like EMPIAR (Electron Microscopy Public Image Archive) [9, 76, 211, 278]. As the SBF-SEM software ecosystem continues to mature, the seamless integration of acquisition metadata, deep learning segmentation architectures, and cloud-based analytical sharing will be paramount to fully deciphering the ultrastructural architecture of biological systems.