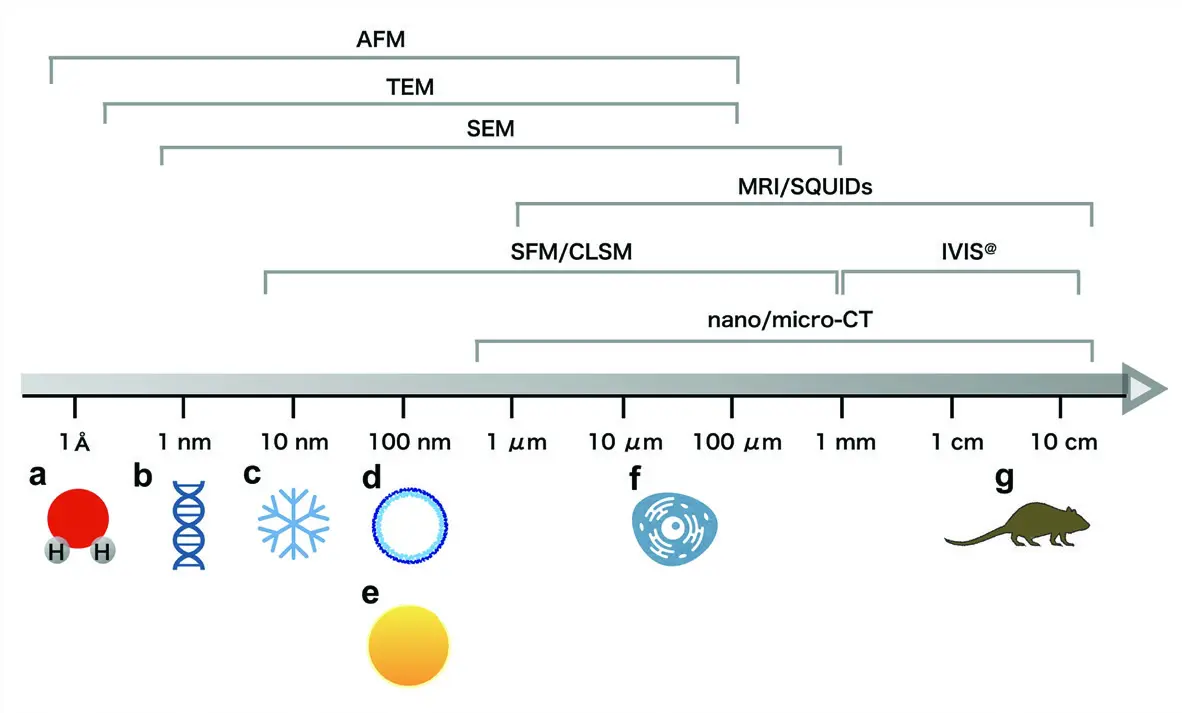
Introduction: The Need for Volume Electron Microscopy
The Complexity of Three-Dimensional Biological Architecture
Living systems are inherently three-dimensional, characterised by complex, hierarchical, and highly graded structures that are intimately linked to their biological functions [9, 23, 38]. Since the widespread adoption of the electron microscope in the mid-20th century, transmission electron microscopy (TEM) has served as the gold standard for structural biology, providing researchers with the unparalleled ability to resolve the nanoscale ultrastructure of cells, tissues, and organelles [3, 10, 20, 28, 37]. With resolutions capable of delineating individual phospholipid bilayers, TEM has immensely contributed to our understanding of cellular networks, cell-cell interactions, and matrix biology [3, 20, 21].
However, conventional TEM is fundamentally a two-dimensional (2D) imaging modality [5, 13, 18, 21, 28]. It relies on the transmission of electrons through exceptionally thin slices of plastic-embedded tissue, typically ranging from 20 to 100 nanometres in thickness [3, 13, 20, 35, 38]. Extracting meaningful three-dimensional (3D) geometric information from these isolated, flat projections is notoriously difficult [10, 28, 35, 38]. Because cells and their constituent organelles—such as convoluted mitochondrial networks, tortuous dendritic spines, and expansive endoplasmic reticula—extend over relatively large volumes, relying on a single 2D cross-section provides only a fragmented snapshot of a much larger architectural reality [6, 28, 32, 38]. Attempting to infer complex 3D relationships, structural interconnectivities, and spatial orientations from these limited 2D views is highly prone to misinterpretation [28, 32, 38]. Consequently, there has been a longstanding and critical need within the life sciences for robust volumetric electron microscopy methodologies capable of bridging the gap between whole-tissue light microscopy and high-resolution ultrastructural analysis [1, 11, 12, 13, 31, 32] (Figure 1).

The Limitations of Traditional Three-Dimensional Electron Microscopy
Prior to the 21st century, researchers seeking to capture 3D ultrastructural data relied almost exclusively on two technically demanding transmission-based methods: electron tomography (ET) and manual serial section TEM (ssTEM) [3, 18, 20, 27, 34]. While both techniques remain powerful tools, they possess inherent limitations that restrict their application for large-scale volumetric studies.
Electron tomography generates 3D reconstructions by physically tilting a relatively thick resin section (typically up to 250 nm) across a wide range of angles—often ±70° within the microscope goniometer—and acquiring a series of projection images at incremental tilt angles [3, 5, 17, 18, 20, 21, 34]. These tilt series are then computationally back-projected to reconstruct a high-resolution tomogram [5, 17, 18, 20, 34]. ET provides exquisite, near-atomic or sub-nanometre spatial resolution, making it ideal for elucidating the architecture of individual macromolecular assemblies, viral particles, or highly localized sub-cellular compartments [13, 20, 32, 34]. However, the technique is fundamentally constrained by its minimal field of view (often just a few micrometres) and extremely shallow depth penetration [5, 13, 18, 20, 21, 33, 34]. As a result, ET is impractical for visualising extensive cellular networks, complete eukaryotic cells, or macroscopic tissue interactions [13, 20, 32].
For studies requiring a broader contextual volume, researchers traditionally turned to manual serial section TEM [1, 14, 29, 34]. This classical approach requires a highly skilled ultramicrotomist to meticulously cut ribbons of consecutive ultrathin sections, physically collect them onto delicate copper grids or solid substrates, and image each section sequentially in the TEM [1, 12, 14, 28, 29, 35]. Although ssTEM has yielded monumental achievements—most notably the complete three-dimensional wiring diagram of the *Caenorhabditis elegans* nervous system—it is an excruciatingly tedious, time-consuming, and labour-intensive process [1, 12, 14, 18, 28, 29, 31, 35]. The manual handling of serial sections is highly susceptible to experimental failure; even a highly proficient operator relies on a degree of luck to avoid losing sections, introducing mechanical folds, causing shrinkage, or inducing stretching artefacts [14, 29, 35]. Furthermore, slight variations in section thickness and physical distortions introduced during collection mean that computationally aligning (registering) the resulting images into an undistorted, coherent 3D stack requires immense manual intervention and complex corrective software [6, 11, 27, 28, 31, 35]. For large volumes, such as those required in mammalian connectomics, the manual sectioning and alignment process can span months or even years, rendering high-throughput studies virtually impossible [11, 16, 28, 31, 35].
The Emergence of Volume Scanning Electron Microscopy
To circumvent the severe throughput bottlenecks and physical vulnerabilities of manual sectioning, the electron microscopy community underwent a paradigm shift, increasingly turning to scanning electron microscopy (SEM) platforms to generate volumetric data [2, 9, 10, 16, 28]. This transition gave rise to a suite of automated technologies collectively termed volume electron microscopy (vEM) [3, 9, 10, 14, 16, 28].
Traditionally, SEM was relegated to imaging the topographical surface of biological or material specimens via the detection of secondary electrons (SEs) under high vacuum [2, 4, 15, 22, 35]. However, modern field-emission SEMs (FE-SEMs) equipped with highly sensitive backscattered electron (BSE) detectors opened a new frontier for biological imaging [2, 6, 22]. The emission of backscattered electrons is strongly dependent on the atomic number (Z) of the interacting material; heavier elements scatter electrons much more efficiently than lighter ones [2, 14, 32, 42]. By deeply impregnating plastic-embedded tissues with heavy metal stains—such as osmium, uranium, and lead—researchers could use the SEM's BSE detector to image the exposed, flat surface of a resin block [1, 2, 6, 13, 14, 17, 42]. The resulting block-face images, when contrast-inverted, provide high-resolution, ultra-structural maps that are remarkably comparable to conventional TEM micrographs, clearly delineating membranes, organelles, and synaptic vesicles without the need to physically transmit electrons through a section [1, 2, 6, 13, 17, 33, 40].
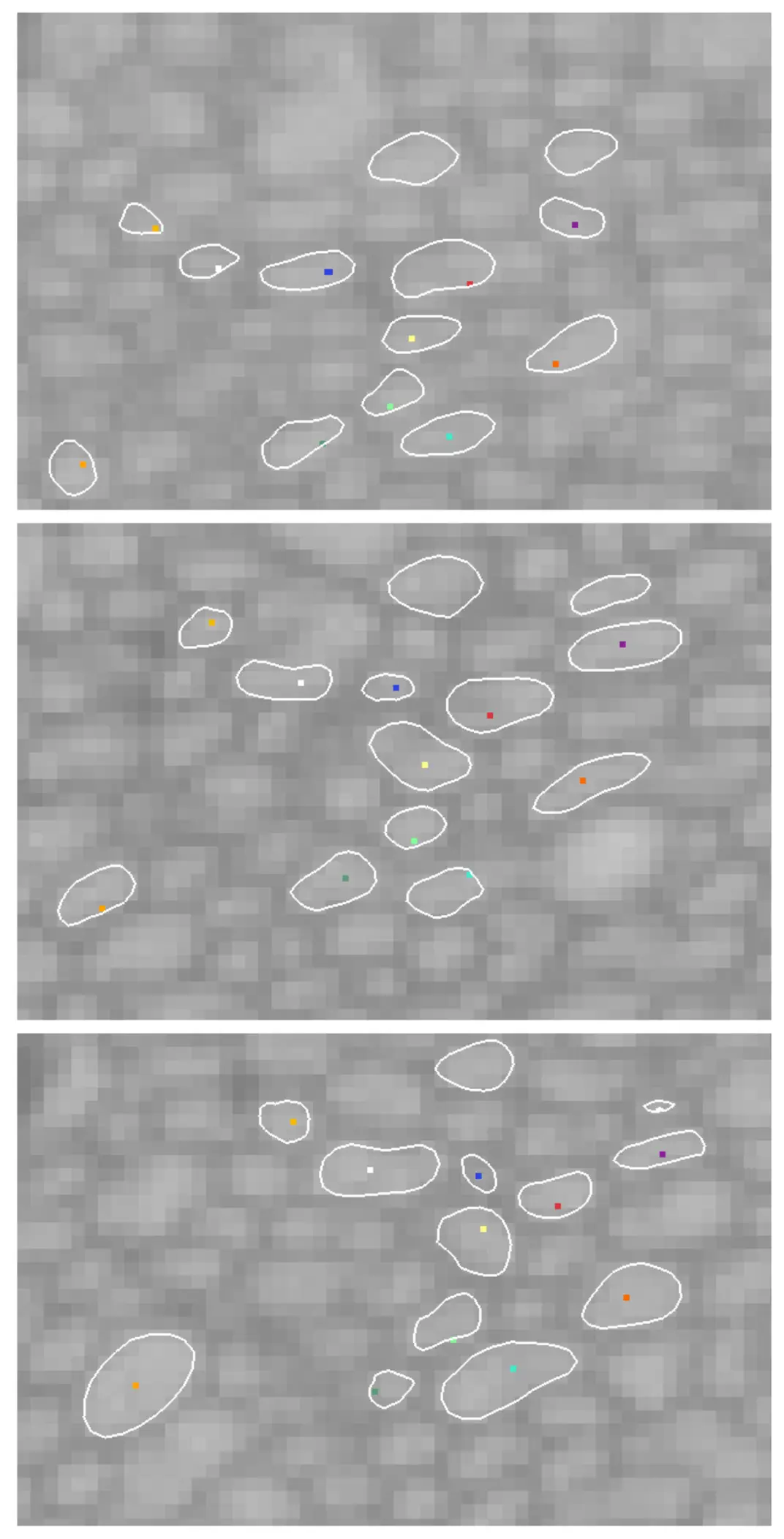
This block-face imaging capability laid the foundation for automated "slice-and-view" vEM techniques [3, 5, 14, 27, 46]. The two most prominent of these are focused ion beam scanning electron microscopy (FIB-SEM) and serial block-face scanning electron microscopy (SBF-SEM) [2, 3, 4, 14, 27, 38]. While FIB-SEM utilizes a heavy ion beam (such as gallium) to precisely mill away nanometre-thick layers from the block surface—yielding isotropic resolutions often below 5 nm—it is generally limited to relatively small tissue volumes (tens of micrometres) due to slow milling speeds [3, 4, 5, 9, 21, 27, 38]. SBF-SEM, on the other hand, employs an automated mechanical ultramicrotome directly within the SEM chamber, sacrificing some axial (Z-axis) resolution in exchange for the rapid, reliable acquisition of vastly larger fields of view (hundreds of micrometres) [1, 4, 5, 20, 28, 32, 34, 38] (Figures 2–5).

Serial Block-Face Scanning Electron Microscopy: A Technological Milestone
The conceptual architecture for SBF-SEM was first introduced in 1981 by Stephen Leighton and Alan Kuzirian, who successfully constructed a prototype miniature ultramicrotome that functioned within the vacuum chamber of a scanning electron microscope [5, 8, 12, 17, 18, 19, 20, 22, 28, 34, 35, 46, 48]. Leighton’s system proved that an embedded tissue block could be mechanically sectioned in situ, with the freshly cut block-face imaged by the electron beam, thus entirely eliminating the need to collect and handle fragile ultrathin sections [12, 17, 28, 34].
However, Leighton's pioneering design was severely hindered by the technological limitations of the era [12, 18, 20, 28, 46]. Standard epoxy resins are highly insulative, meaning that the electron beam rapidly deposits a negative charge on the sample surface [1, 20, 29]. In a traditional high-vacuum SEM, this surface charging severely deflects the primary electron beam, drastically degrading image resolution and inducing massive distortions [1, 20, 29, 36]. To counteract this in the 1980s, the block-face had to be removed from the vacuum chamber and sputter-coated with a conductive layer of carbon or gold between every single cut, rendering the process agonizingly slow and negating the benefits of automation [18, 20, 26]. Furthermore, the computing power and digital storage necessary to manage and reconstruct thousands of high-resolution digital micrographs simply did not exist [12, 18, 20, 26, 46]. Consequently, the technique was largely abandoned for over two decades [12, 18, 46].
The technique was spectacularly resurrected and refined in 2004 by physicists Winfried Denk and Heinz Horstmann at the Max Planck Institute for Medical Research [2, 3, 5, 8, 11, 12, 13, 17, 18, 20, 22, 26, 28, 32, 34, 35, 37, 46, 48, 49]. Denk and Horstmann introduced a pivotal innovation that solved the charging problem without the need for repetitive metallic coating: they utilized an SEM operating in variable pressure (or low-vacuum) mode [12, 13, 18, 20]. By bleeding a precisely controlled, minute amount of water vapour or nitrogen gas (e.g., 20–60 Pa) into the specimen chamber, the primary electron beam ionises the gas molecules [13, 18, 20, 47]. These positive ions rain down onto the specimen surface, effectively neutralising the accumulated negative charge on the uncoated block-face [13, 20, 26].
Operating in this low-vacuum environment, Denk and Horstmann demonstrated that their automated microtome could iteratively shave 50–70 nm slices from a block of neural tissue while capturing sequentially perfectly aligned BSE images [11, 13]. Because the sample remains physically stationary in the lateral (X-Y) plane during the cutting and imaging cycle, the resulting image stacks possess near-perfect inherent registration, eliminating the severe computational alignment bottlenecks associated with manual ssTEM [6, 7, 14, 31, 36, 41]. The lateral resolution achieved was sufficient to track the finest axons and identify individual synaptic vesicles [11, 13].
Recognizing the transformative potential of this setup, the technology was subsequently commercialised by Gatan Inc. as the "3View" system, a module that replaces the standard door of a host SEM [1, 6, 12, 17, 18, 20, 22, 34, 35, 37, 46]. As the demand for vEM exploded, other manufacturers entered the space, yielding integrated systems such as the Thermo Fisher Teneo Volumescope and the ConnectomX Katana [1, 17, 18, 19, 20, 22, 51]. Modern SBF-SEM platforms utilise oscillating diamond knives to minimize physical compression and damage during sectioning, enabling highly reliable slicing at thicknesses commonly between 25 and 50 nm [4, 11, 38, 41, 50]. With automated data collection capable of running continuously for days or weeks, SBF-SEM firmly established itself as an off-the-shelf, high-throughput solution for acquiring massive 3D ultrastructural datasets encompassing millions of cubic micrometres [12, 13, 14, 31, 32, 42].
Overcoming Sample Preparation Hurdles: En Bloc Staining and Conductivity
While SBF-SEM hardware revolutionized image acquisition, the transition from imaging ultra-thin physical sections to imaging the face of a bulk plastic block necessitated a fundamental overhaul in biological sample preparation [8, 11, 29, 38, 44]. In conventional TEM, contrast is often enhanced by post-staining individual sections on a grid. In SBF-SEM, because the microtome continuously exposes fresh layers of tissue deep within the block, the entire specimen must be thoroughly and uniformly contrasted *en bloc* before it is dehydrated and infiltrated with resin [8, 11, 14, 18, 22, 44].
Furthermore, despite the mitigating effects of variable pressure SEM, the imaging of highly insulative epoxy resins still poses severe charging risks, particularly at the low accelerating voltages (e.g., 1–3 keV) required to achieve high Z-axis resolution [1, 29, 36, 38, 42, 44]. High beam energies would penetrate too deeply into the sample block (up to several micrometres), causing subsurface scattering that blurs structural boundaries and destroys the axial resolution [19, 25, 42]. Conversely, operating at very low beam energies requires a highly robust BSE signal to generate a viable image with an acceptable signal-to-noise ratio [1, 25, 42, 44]. Additionally, operating the SEM at higher chamber pressures to reduce charging has the adverse effect of scattering the primary electron beam (the "skirt" effect), which inherently reduces lateral spatial resolution [1, 45].
To overcome these conflicting physical limitations, the vEM community developed aggressive, multi-step heavy metal staining protocols designed to simultaneously maximize electron backscattering and vastly increase the internal electrical conductivity of the biological sample [1, 4, 8, 14, 17, 18, 22, 44]. The most widely adopted framework is the National Center for Microscopy and Imaging Research (NCMIR) protocol [1, 8, 10, 11, 14, 34]. This rigorous methodology utilizes an osmium-thiocarbohydrazide-osmium (OTO) sequence, which leverages thiocarbohydrazide as a mordant to bridge and bind secondary layers of osmium tetroxide directly onto cellular lipid membranes [1, 12, 52]. This is followed by *en bloc* incubation in uranyl acetate and *en bloc* lead aspartate [1, 12, 52]. The massive deposition of heavy metals transforms the cellular architecture into a highly conductive, electron-dense scaffold, effectively turning the biological tissue into a pseudo-metallic network [1, 8, 44].
Such intensive contrasting protocols ensure that the low-energy primary electron beam interacts almost exclusively with the uppermost 20 to 30 nanometres of the block-face [25, 42]. This shallow interaction volume is critical, as it aligns the imaging depth with the physical slice thickness of the microtome, allowing for precise 3D rendering without subsurface blurring [19, 25, 32, 42]. In challenging specimens where charging remains problematic, researchers have also implemented conductive resins spiked with carbon black (Ketjenblack) or utilized minimalistic embedding techniques that wash away excess insulating resin from the periphery of the sample prior to polymerization [4, 12, 18, 29].
Bridging Specificity and Structure: SBF-SEM and CLEM
Initially championed by neuroscientists to chart the dense, complex wiring diagrams of the brain (connectomics), the exceptional utility of SBF-SEM has rapidly transcended its neurological origins [10, 11, 12, 16, 17, 20, 26, 29, 32]. Today, the technique is widely deployed across diverse biological disciplines, illuminating everything from the ultrastructural remodelling of mitochondria in cancer cells and the 3D architecture of kidney podocytes, to the intricate infection mechanisms of parasites and the spatial organisation of plant cell extracellular matrices [10, 11, 12, 17, 26, 29, 32, 39, 40, 47].
However, a fundamental limitation of all volume electron microscopy, including SBF-SEM, is its lack of molecular specificity [24]. While the technique provides a breathtaking, pan-cellular view of cellular topography and membrane morphology, it relies entirely on non-specific heavy metal stains, making it impossible to definitively identify specific protein populations, genetic targets, or transient molecular events solely from the greyscale EM micrographs [24, 30].
To resolve this limitation, SBF-SEM is increasingly integrated into Correlative Light and Electron Microscopy (CLEM) workflows [2, 3, 4, 5, 14, 22, 26, 30, 46, 50]. CLEM elegantly bridges the macro- and micro-worlds by superimposing the dynamic tracking and molecular precision of fluorescence light microscopy (LM) onto the high-resolution architectural map generated by vEM [3, 14, 22, 30, 46].
In a typical vCLEM workflow, researchers utilise confocal or multi-photon fluorescence microscopy to observe a living biological system—such as transient blood vessel fusion, rare cell division events, or specific fluorescently-tagged organelles [2, 5, 26, 28, 50]. Once the rare or specifically targeted event is captured optically, the sample is rapidly fixed to preserve the exact physiological state [26]. Because searching blindly for a singular, microscopic event within a massive, opaque resin block using EM alone is virtually impossible, researchers utilise the fluorescence data as a 3D navigational map [5, 26, 28, 30, 50]. Precise localization is often aided by burning fiducial marks into the tissue using near-infrared branding (NIRB) lasers during the LM step [50]. These permanent physical scars remain visible after heavy metal staining and resin embedding, serving as coordinates to guide the SBF-SEM microtome directly to the targeted region of interest (ROI) [50].
By combining the targeted molecular identification of LM with the automated, large-volume ultrastructural sectioning of SBF-SEM, researchers can achieve highly efficient, targeted 3D reconstructions of exceedingly rare cellular phenomena [2, 5, 26, 28, 30, 50]. This synergy not only vastly reduces the time wasted imaging irrelevant tissue but also ensures that the computationally heavy and laborious task of 3D image segmentation is focused precisely on biologically meaningful structures [5, 26, 30]. Ultimately, the evolution from manual serial sectioning to automated SBF-SEM—and its subsequent integration with CLEM—represents a monumental leap in structural biology, providing researchers with an unprecedented window into the intricate, three-dimensional spatial relationships that govern life at the nanoscale [3, 9, 14, 16, 18, 43].